YAN Wen-Li LIANG Zhen YU Xing-Lian ZHANG Rong
(School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, China)
ABSTRACT The further interaction mechanism towards renin inhibitors was revealed by comparison of renin with different active inhibitors in aqueous solution. Molecular docking and molecular dynamics (MD)simulations were combined for the research. The results reflected that electrostatic and hydrophobic effects were the major interactions for renin inhibitors forming complexes with renin, and some residues were the key to the formation of complex, especially Asp38/Asp226. The factor of different activities performed in renin inhibitors was illustrated as well. For the higher active renin inhibitor, it possessed stronger affinity with renin, and its detected conformation was more extended to fit for the key binding site. This promoted the capacity to form special interactions with the key residues. While conformation of the lower active renin inhibitor performed folded in the active site of renin, the interactions to the important pocket S3sp was restricted, resulting in undesirable bioactivity.
Keywords: renin inhibitors, mechanism, molecular docking, molecular dynamics (MD) simulations;
RAS (renin-angiotensin system) is an important endocrine system for regulating blood pressure and fluid homeostasis,playing a vital role in the internal environment[1-3]. The overactivation of RAS induces excessive generation of angiotensin II, which leads to the cardiovascular disease[4,5].The common drugs for the regulation of RAS are angiotensin I converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs) and renin inhibitors (RIs)[6-8]. However,ACEIs and ARBs will not totally block the RAS. RIs are considered more effective to prevent and reverse the damage of the target organ, inhibiting renin directly at the ratelimiting step[9,10]. Based on the structure-based drug design,the first approved nonpeptide renin inhibitor aliskiren was discovered, which possessed the ability to interact with the hydrophobic S1/S3-binding pocket of renin[11]. With the success of aliskiren, the study of renin inhibitors attracted widespread attention[12,13]. Many researchers have recognized the significance of structure-activity relationship and the action mechanism in renin inhibitors[14-16], which is the key to design new drugs. As some side effects exist in aliskiren[17,18],it is still necessary to find better renin inhibitors.
Computer-aided drug design (CADD) as a brand new technique for drug discovery is widely used in various pharmaceutical researches[19]. The advancement in CADD enables novel techniques for renin inhibitors to explore the action mechanism[20]. Many minuscule mechanism details of pharmacology were revealed by the method[21-24]. Molecular docking and molecular dynamics (MD) simulation are the principal approaches to calculate the interaction between the drug and targets[25-27]. In recent years, the methods have been widely applied for analyzing rational conformation between drug and its receptor, which promotes the theory discussion of internal mechanism[28-31]. As the renin inhibitors were reported with five major active pockets for bounding to the renin[11], it was essential to understand the details between renin inhibitors and active pockets. Herein, by means of molecular docking and molecular dynamics simulation, two homologous renin inhibitors in different activities were applied to observe the combinative disparity with renin on the molecule level.

Table 1. Component of the Pockets of Renin
2. 1 Molecular docking
The molecules of high activity (aliskiren) and low activity(compound 1) were chosen as the ligands, which were constructed by Chem3D module of Chemoffice, and their information is exhibited in Fig. 1. Energies of the ligands were auto minimized in the MMFF94 force field for the next docking. The X-ray crystal of renin (ID: 2v10, resembling the common skeleton in different active renin inhibitors) was used as the receptor. Extra crystalline water molecule and unrelated ions such as Na+and CL-of renin were removed by Pymol 2.3.0 software. AutoDock 4.2 was applied for calculating the binding mode of renin and renin inhibitor. The ligands and receptors added with hydrogen and charges were saved as the format pdbqt for preparation. And the grid box was set as a cube with the length of 1.5 Å. The docking was executed in the form of semi-flexibility with the rigid receptor and flexible ligand. And the optimal binding modes of ligand and receptor were screen out by Lamarckian genetic algorithm.
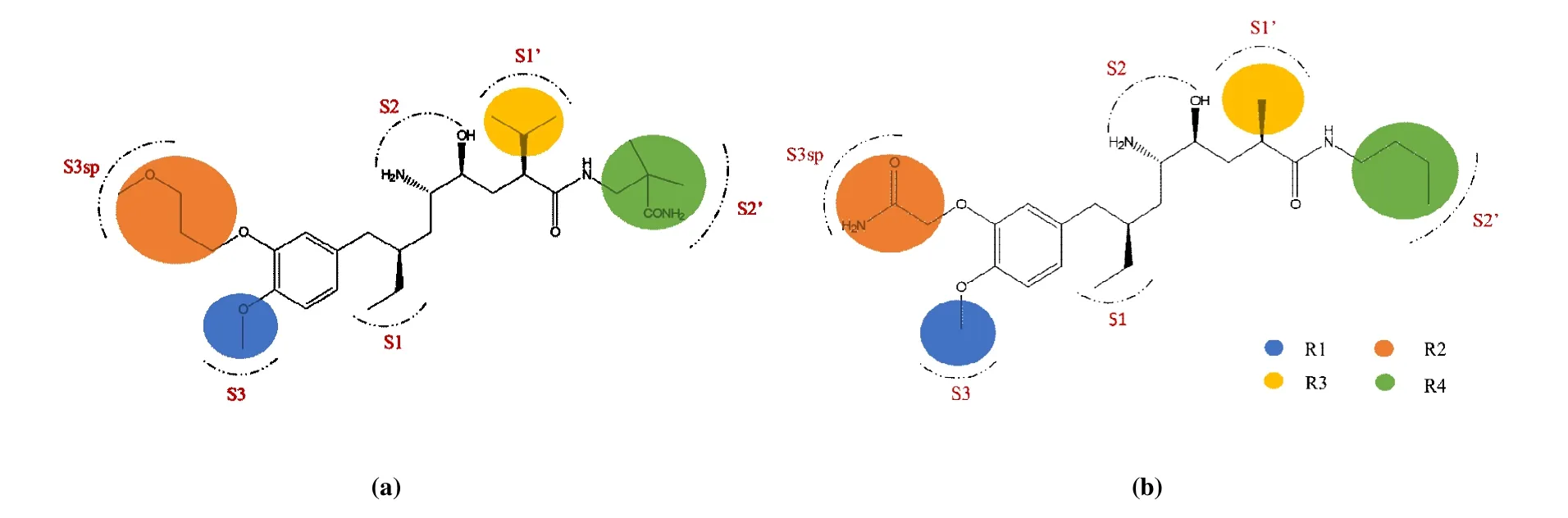
Fig. 1. Structures and pocket distributions of aliskiren (a) and compound 1 (b)
2. 2 Molecular dynamics simulation
MD simulations were adopted to study the mechanism between renin and the different inhibitors by GROMACS 5.1.2 package. Gromacs96 force filed was used for the receptor and ligands[32]. The simulation was carried out in a cubic type of box filled with SPC water, and Na+and CLwere added for neutralizing the system[33]. Canonical NVT(isothermal-isochoric ensemble) and NPT (isothermalisobaric ensemble) with periodic boundary conditions were set for the equilibration. The temperature and pressure were kept constant at 300 and 101.3 kPa, respectively. Both NPT and NVT of equilibration were performed for 100 ps. Cut-off distances for the calculation of Coulomb and van der Waals interactions were set as 1.0 nm. Finally, 25 ns simulation was continued with the result of 5 ns simulation as the initial conformation. The simulation conditions for both two complexes (renin-compound 1 and aliskiren) and renin in the absence of ligand were the same.
3. 1 Result of molecular docking
Optimal conformations for complexes of receptor and ligand (renin-aliskiren and renin-compound 1) were screened out. The highly active ligand aliskiren was obtained with a docking energy value of -8.30 kJ /mol. And the lowly active compound 1 was obtained by the same operation with an energy value of -6.08 kJ/mol. Conformations of the two inhibitors in comparison with the prime ligand of renin are shown in Fig. 2. The docking results denoted the objects corresponding to the way of original ligand bound in the renin crystal. And the interactions of the docked ligand with renin are explicated in Fig. 3.
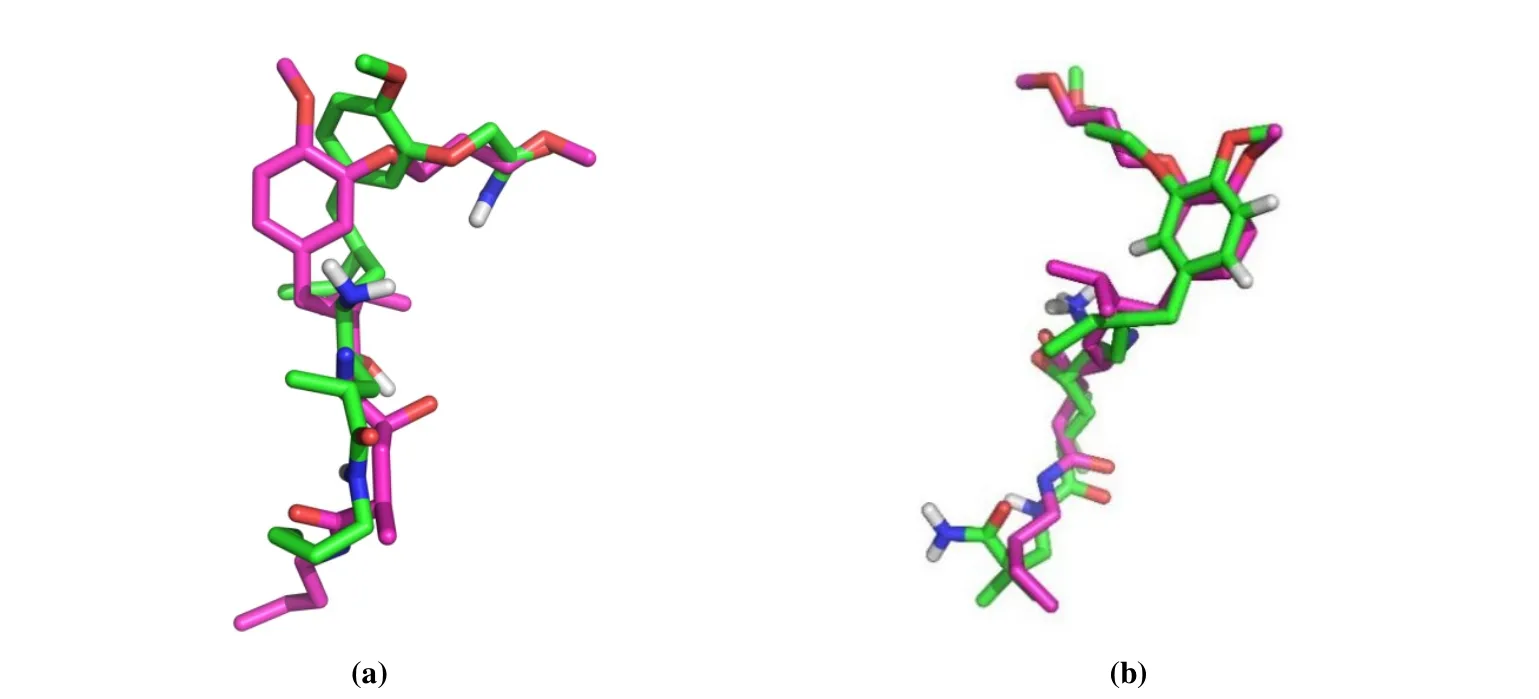
Fig. 2. Comparisons of the docking results (green) and original ligand (purple) of renin: aliskiren (a) and compound 1 (b)

Fig. 3. Specific interactions of renin with aliskiren (a) and compound 1 (b)
3. 2 Result of molecular dynamics simulation
3. 2. 1 Landscape of the combination
In the molecular dynamics simulation, the root-meansquare deviation (RMSD) trajectory was performed to investigate the stability of the complexes (renin-aliskiren and renin-compound 1). As Fig. 4 shows, theRMSDof renin combined with aliskiren or compound 1 reached equilibrium faster than the renin in the absence of ligands. And the variation inRMSDof renin binding with ligand is more stable than renin without ligand, indicating that the stabilization of renin would be enhanced in the presence of renin inhibitors.
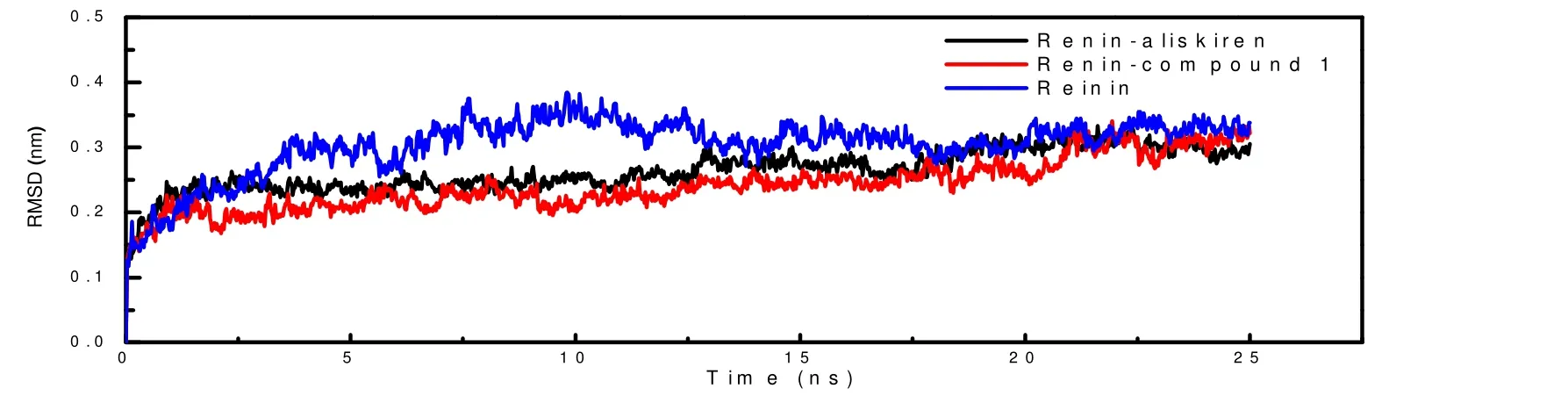
Fig. 4. RMSDs of the complexes and purified renin
To find out more information of the interaction and combination between renin and renin inhibitors, the binding energy was calculated by g_mmpbsa, a plugin of Gromacs software, with the results performed in Table 2. In solution,renin inhibitors bound to renin were mainly mediated by van der Waals force, while the role of electrostatic force was very faint. As described in Table 2, the total binding energies of renin-aliskiren and renin-compound 1 were respectively at-271.78 and -221.22 kJ/mol, demonstrating that the affinity of renin and aliskiren was stronger, in accordance with the high experimental pIC50of aliskiren.

Table 2. Binding Energy of Renin-aliskiren and Renin-compound 1
Theb-factor (temperature factor) in Fig. 5 was introduced to describe the details of overall combination. The mean structures of renin, renin-aliskiren and renin-compound 1 were calculated to investigate theb-factor. In Fig. 5, the stabilization was described by colors, and the extents of stability in proper order were blue, green, yellow and red.Blue represents the most stable conformation and red refers to the most active conformation. According to theb-factor, most residues of the complexes renin-aliskiren and renin-compound 1 are blue, representing that the structures are stable (Figs. 5a and 5b). However, in the case of the renin without ligand,there are many other colors except blue (Fig. 5c). And in the active pockets, this phenomenon was prominent. Apparently,contrasting with the renin absent of ligands, the stability of renin was promoted by the renin inhibitors.

Fig. 5. B-factors of purified renin (a) and the complex renin with renin-aliskiren (b) and renin-compound 1(c)

Fig. 6. Covariance matrix of α-C atom of purified renin (a) or the complex renin with renin-aliskiren (b) or renin-compound 1 (c)
The covariance matrix ofα-Catom in renin residues based on 2-dimension principal component analyses (PCA) was performed in Fig. 6. It was applied for describing the intensive motion of particles. In the covariance matrix, red indicated that two atoms were moving towards the same direction, while blue was on the opposite. By comparing with the renin without ligand in Fig. 6a, the color depth of the blue decreased to some extent in Fig. 6b and Fig. 6c. This suggested the renin inhibitors would reduce irrelevance between renin residues and promote stability of renin due to the presence of van der Waals forces generated by ligands.
3. 2. 2 Details of the binding sites
In order to detect the specific details of the combination between renin and renin inhibitors, the interactions of renin inhibitors in active pockets of renin were investigated. The important binding pockets (S1, S1’, S2’, S3, S3sp) for the renin inhibitors had been described by Rahuel et al, and the components of the pockets are listed in Table 1[11]. The hydrophobic interactions had influence on the inhibitors binding to the active sites. As depicted in Fig. 7a, aliskiren was restricted to a large hydrophobic pocket consist of the key binding site S1, S1’, S2’, S3 and S3sp. Compound 1 was in the hydrophobic pocket formed by S1, S1’, S3 and S3sp in Fig. 7b. Distinctly, aliskiren occupied more binding sites than compound 1 due to the larger size. Comparison for the interactions of the two different ligands is shown in Fig. 8.The residues surrounded with the ligand at range of 6 Å were described as ribbons, and the ligands are shown as sticks in Figs. 8a and 8c. According to Fig. 8, the conformation of aliskiren was more extended than that of compound 1.Therefore, there was more opportunity for aliskiren to contact with the key residues of renin. And compared with compound 1, the orientation of aliskiren appeared better to fit the surface of the hydrophobic pockets.

Fig. 7. Hydrophobic interactions of renin in complex with aliskiren (a) and compound 1 (b)

Fig. 8. General view of aliskiren (a) or compound 1 (c) with residues in renin within 6 Å. Specific interactions between renin and higher active compound 26 (b) or lower active compound 1 (d)
A higher resolution scan of the interaction between renin and renin inhibitors was demonstrated in Figs. 8b and 8d. As shown in the images, the binding occurred mostly through weak non-covalent interactions, such as van der Waals force and hydrogen bonding. Among the forces, van der Waals force was the major driving forces. In Figs. 8b and 8d, more interactions were found between renin and aliskiren than that between renin with compound 1. According to Rahuel, the interaction with S1/S3-binding pocket in renin promised the vitro activity. Besides, the additional hydrogen bonding or hydrophobic effects with binding site S3sp of inhibitors were essential for the higher bioactivity[11]. Exactly, the result of MD simulation was matched with the rule. Both the highly and lowly active renin inhibitors could interact with S1, but only aliskiren formed hydrogen bonding with the S3sp. As Fig. 8b shows, aliskiren possessed electrostatic interactions with pocket S3sp (Gly228), S2’ (Gln135, Gly40) and S1(Asp226, Asp38), conjugation effect with S1 (Phe124, Tyr83,Val127), S1’ (Leu224), S2’ (Ile137) and S3sp (Val127) and van der Waals force with all the pockets above. However,compound 1 possessed electrostatic and conjugate interactions with S1 (Tyr83 and Thr85) and S3 (Pro118, Phe119, Ala122 and Phe124) merely (Fig. 8d), and the van der Waals interactions of compound 1 were less than that of aliskiren.Apparently, 1 was not optimistic to interact with the important pocket, thus resulting in the undesired inhibitory activity.
In addition, Asp32 and Asp 215 were imperative for catalysis of renin. Occupying the sites was favor for inhibiting activity of renin inhibitors. The binding energy variation of the two inhibitors with Asp38 and Asp 226 was extracted in Fig. 9. As it depicted, aliskiren possessed more stable and powerful binding energy with the two key residues than compound 1. This reveals the fact that compound 1 is not effective enough to meet the interactive needs of renin.

Fig. 9. Variations of different active renin inhibitors binding with Asp32 (a) and Asp215 (b)
The binding energy contribution in Fig. 10 revealed the role of residues to the combination. In the complex of reninaliskiren, residues Gln19, Tyr83, Thr85 and Phe124 made the significant contribution to the binding. And in the complex of renin-compound 1, Arg82, Tyr83 and Phe124 were vital for the combination. The contribution of residue Tyr83 and Phe124 in pocket S1 was prominent in both the two complexes. It proved that pocket S1 was important for the bioactivity of renin inhibitors as well.

Fig. 10. Binding energy contribution of complex renin-compound 26 (a) and renin-compound 1 (b)
The match manners of renin and renin inhibitors were detected by molecular docking in the study. For more insight,the highly active inhibitor aliskiren and the lowest active compound 1 were compared in aqueous solution through theRMSD, binding energy,b-factor, PCA and internal interactions by MD simulations. The mechanism of different inhibitors exhibiting diverse activity has been revealed. The results ofRMSD,b-factor and PCA confirmed that the stabilization of renin could be promoted in the presence of the ligands. The binding energy revealed that the combination of renin and renin inhibitors was mainly mediated by van der Waals force. And the higher active inhibitor had stronger binding ability with key residues Asp38 and Asp226. The calculated interactions demonstrated that both the renin inhibitors interacted with a large hydrophobic pocket of renin,and the structure of higher active inhibitor was more extended to access to interact with the residues in the binding sites. The interaction of renin inhibitor with S1 and S3 binding site guaranteed the bioactivity, and the additional hydrogenbonding interactions interaction to S3sp could enhance the activity. The results help us comprehend the mechanism of renin inhibitor more profoundly.