李灵梅 魏亿芳 李 治 房瑞玲 崔跃华 曹红艳,4△
【提 要】 目的 探讨局部保留投影的正则化多核学习(regularized multiple kernel learning with locality preserving projections,rMKL-LPP)在乳头状肾细胞癌(papillary renal cell carcinoma,PRCC)多组学数据分子分型中的应用,进一步研究PRCC分子分型在信号通路活性和基因表达调控方面的异质性。方法 采用rMKL-LPP方法对PRCC的mRNA、miRNA和DNA甲基化数据进行整合,进一步采用k-means方法聚类分型,并通过Cox回归分析研究不同分型的预后风险。针对不同分型,进行通路活性分析,使用差异表达分析筛选DEmRNAs(differentially expressed mRNAs),DEmiRNAs(differentially expressed miRNAs)和DMGs(differentially methylated genes),并对三者的重合基因进行GO(gene ontology)富集分析,最后使用相关及生存分析筛选可能受DNA甲基化或miRNA调控且影响患者生存的基因。结果 PRCC患者分为三型,不同亚型在通路活性和基因表达方面均有差异。筛选出10条活性存在差异的通路;
1185个DEmRNAs,13个DEmiRNAs及416个DMGs,其中36个重合基因富集于有统计学差异的8个GO生物项。相关分析发现,ABL2可能受hsa-miR-107调控,13个基因可能受DNA甲基化调控。生存分析表明,ZNF135和RBPMS2可能与患者生存结局相关。结论 rMKL-LPP能够有效识别PRCC亚型,筛选出的通路及潜在生物标志物,可为PRCC针对性治疗提供依据。
乳头状肾细胞癌(papillary renal cell carcinoma,PRCC)是肾细胞癌(renal cell carcinoma,RCC)的第二常见亚型,占RCC病例的10%~15%[1]。PRCC具有很强的异质性,组织学上通常分为两型:Type Ⅰ和Type Ⅱ。其中,Type Ⅰ型属于低级别肿瘤,Type Ⅱ型属于高级别肿瘤[2]。Type Ⅱ较Type Ⅰ异质性更强,预后更差[3],可分化为高度恶性的RCC肉瘤样型[4]。该组织学分型常用于传统临床对PRCC患者进行预后评估,然而同一组织类型且临床分期相近的患者即使采用相同的治疗手段,其治疗效果和预后结局亦相差较大,评估效果并不理想。随着测序技术的发展,积累了大量组学数据,从组学层面研究癌症分子分型随之兴起。整合多组学数据不仅能够捕获PRCC不同组学的异质性,同时还可获得组学间的关联信息[5],从多层面揭示疾病的复杂调控机制。如何利用组学数据对患者精准分型,为治疗方案的选择及预后评估提供帮助,进而实现精准治疗,是PRCC临床治疗的重要发展方向。
TCGA研究组[3](2016)采用COCA方法[6](cluster-of-clusters analysis)综合PRCC患者的miRNA/mRNA、拷贝数变异、蛋白表达数据及DNA甲基化数据,首次对PRCC患者进行了分子分型。COCA是一种两步聚类法,首先基于不同数据类型的聚类结果构建一个二进制矩阵,然后输入该矩阵进行一致性聚类,得到一个综合不同数据集的全局聚类结构。然而,COCA属于后期整合方法,在对每个组学数据进行单独聚类时,易损失较弱的数据信号[7],而且在组合不同数据的聚类结构时未能考虑不同组学对分型的贡献[8]。而基于多核学习[9]的方法,将不同n×pi的组学数据分别转换为n×n的样本相似矩阵,通过学习优化,得到最优样本相似矩阵的线性组合,能够反映不同类型数据的权重,在多组学数据整合分型中独具优势。Speicher等[10]将多核学习与局部保留投影降维方法[11](locality preserving projections,LPP)结合,提出了rMKL-LPP。rMKL-LPP具有以下特点:(1)可基于数据类型灵活选择核函数;
(2)样本相似矩阵的权重即为不同组学数据的贡献度,反映了不同组学对分型的贡献;
(3)每个数据类型可设置多个核函数,避免了核参数设定的局限性。此外,Rappoport和Shamir[7]研究不同整合方法在10种TCGA癌症分型中的应用时,指出rMKL-LPP相比其他方法,更能有效识别出与临床特征及生存率显著相关的分子亚型。
因此,本文采用rMKL-LPP算法,整合PRCC患者mRNA、miRNA及DNA甲基化数据进行分型,并寻找不同分型的重要通路及差异表达基因,为实现乳头状肾细胞癌不同分型的针对性治疗提供参考。
1.数据来源
使用R包TCGAbiolinks[12]下载PRCC的mRNA、miRNA、DNA甲基化及临床数据,进行ID匹配后,得到表达矩阵:56493×219的mRNA矩阵、1881×219的miRNA矩阵及485577×219的DNA甲基化矩阵,其中行表示每个组学数据的特征,列表示样本。数据预处理方法如下:(1)对启动子区域CpG甲基化位点进行注释,启动子区域为转录起始位点2kbp内的区域[13],进一步去除性染色体上的启动子区CpG位点。(2)删除缺失比例大于30%的特征,用KNN(k-nearest neighbors)算法填补剩余缺失值,并对mRNA和miRNA数据进行log2转换。最终得到16534个mRNA,437个miRNA和49022个DNA甲基化位点。
2.分析方法
多核学习降维(multiple kernel learning for dimensionality reduction,MKL-DR)方法[14]通过使用核函数,将不同数据集映射到高维空间并进行集成,然后通过降维算法将集成结果映射到低维空间,进行后续分析[15]。rMKL-LPP在MKL-DR的基础上,采用LPP进行降维,同时为了避免优化问题中的过拟合,加入了正则约束项。方法原理如下:
(1)多核学习
多核学习将M个给定的基本核函数{k1,…,kM}线性组合,通过优化权重系数得到一个融合核K,如公式(1)所示。
(1)
其中Km表示基本核函数,βm是核函数Km的权重系数。
(2)局部保留投影降维LPP
LPP是一种基于图嵌入框架的无监督方法,旨在寻找最优投影向量v,使得经v映射后,样本在优化空间中仍然能够保持高维空间中的近邻关系。v根据图保留准则(graph-preserving criterion)进行优化:
(2)
(3)
(4)
(5)
其中xi和xj表示第i和j个样本,元素wij构成相似矩阵W,元素dij组成约束矩阵D,Nk(i)和Nk(j)为数据点i和j的最近邻数。
(3)引入正则约束的优化

(6)
其中α是一维情况下的投影向量,Ki为集成空间,β是核函数的权重向量。对于多维数据,将针对投影矩阵A=[α1…αp]进行优化,并采用坐标下降法交替对A与β进行迭代优化,直到达到收敛或最大迭代次数。若从优化A开始,令所有核矩阵权重β数值相同,且总和为1;
若从优化β开始,AAT应初始化为I。
(4)k-means聚类
rMKL-LPP通过LPP将集成结果投影到低维空间,进一步采用k-means方法进行聚类分型,并根据轮廓系数选择最优分型数。
(5)rMKL-LPP参数选择

3.基于PRCC分型结果的下游分析
(1)Cox回归分析
控制初始诊断年龄、性别和病理诊断分期等情况下,采用Cox回归分析对PRCC患者的分型结果进行预后评估。
(2)通路活性分析
利用progeny软件包[16]对不同亚型进行通路活性分析,并采用非参数检验筛选活性存在差异的通路,筛选标准为Padj<0.01。
(3)差异基因筛选
采用Kruskal-Wallis秩和检验筛选DEmRNAs、DEmiRNAs及DMGs,阈值设为Padj<0.01;
进一步采用超几何分布检验[17]筛选在每个分型上富集的特征,筛选标准为Padj<0.01。为选择最具代表性的特征,要求特征在该分型中至少有2/3的样本发生改变,同时至少在一个其他分型中少于1/3样本发生改变,按此标准选出的特征即为最终的差异基因。
(4)GO富集分析
利用miRWalk[18]在线工具预测DEmiRNAs的靶基因,进一步采用clusterprofile R包[19]对DEmRNAs、DEmiRNAs靶基因及DMGs的重合基因进行富集分析。
(5)相关分析
分别对DEmRNAs与DMGs的重合基因,DEmRNAs与DEmiRNAs靶基因的重合基因进行Pearson相关分析,根据相关系数r和P值筛选出可能受DNA甲基化负调控的基因,筛选标准为-1.0 (6)基因生存分析 对可能受DNA甲基化或miRNA调控的基因进行生存分析。根据基因表达水平将患者分为高表达组和低表达组,应用Kaplan-Meier生存分析筛选与患者生存相关的基因,筛选标准为P<0.05。 1.PRCC患者分型结果评价 采用rMKL-LPP对219名PRCC患者的组学数据进行整合分析,最优分型数为4(图1),生存曲线见图2,不同分型患者的生存率存在差异(χ2=89.566,P<0.0001)。经Cox回归分析发现,Cluster2和Cluster3生存率差异无统计学意义(χ2=0.050,P=0.823)。因此,将Cluster2和Cluster3合并成为一个新的Cluster2,三组基本资料见表1,生存曲线见图3。结果显示,Cluster3患者相比于其他两型,预后差,说明基于rMKL-LPP的分型与PRCC患者生存相关。 图1 PRCC分型结果的三维图 图2 PRCC分为四型的生存曲线图 图3 PRCC分为三型的生存曲线图 表1 PRCC患者分型的基本资料 在校正协变量的情况下,研究不同分型对预后的影响,即分型作为自变量,生存时间和生存状态作为因变量,拟合Cox回归模型,结果如表2,预后最差的Cluster3患者的死亡风险是Cluster1的47.731倍,Cluster2患者的死亡风险是Cluster1的6.143倍; 表2 219例PRCC患者的Cox回归分析结果 2.通路活性分析 对PRCC亚型进行通路活性分析,存在差异的10条通路如图4所示,其中TGF-β、EGFR、NF-Kβ、MAPK、Hypoxia、TNF-α和PI3K通路在Cluster3中活性最高; 图4 PRCC不同亚型的差异通路 3.差异基因筛选及分析 (1)差异基因的筛选结果 筛选出1185个DEmRNAs,其中上调626个,下调559个; 图5 不同亚型中差异基因表达热图 对13个DEmiRNAs进行预测得到36个靶基因,通过对DEmRNAs,DMGs以及36个DEmiRNAs靶基因进行联合分析,发现DEmiRNAs靶基因与DEmRNAs有1个重合基因,DMGs与DEmRNAs有35个重合基因(图6)。 图6 差异基因的韦恩图 (2)GO富集分析 为进一步验证基于rMKL-LPP分型的生物学意义,对联合分析得到的重合基因进行富集分析。36个重合基因富集于8个GO生物项,见图7,基因与GO生物项的关系如图8所示。GO富集分析可从生物过程(biological process,BP)、分子功能(molecular function,MF)和细胞组成(cellular component,CC)等三部分对基因及基因产物进行注释。8个GO生物项主要体现在生物过程和细胞组成两个方面。图7中8个GO生物项纵轴自上而下依次为中胚层发育、色氨酸分解过程、含吲哚化合物分解代谢过程、吲哚烷基胺分解过程、色氨酸代谢过程、吲哚烷基胺代谢过程、顶端质膜与细胞的顶端部分。图中实心圆的大小表示富集于该通路基因的数量。 图7 GO通路分析图 图8 基因与8个GO生物项的网络关系图 (3)相关分析 对DEmRNAs与DMGs重合基因进行相关分析,最终得到13个存在相关关系的基因,即可能受DNA甲基化调控的基因,如图9所示。基因在不同亚型表现出不同的相关关系,如ZNF135仅在Cluster1中存在相关关系; 图9 重合基因相关关系热图 (4)基因生存分析 对可能受DNA甲基化或miRNA调控的基因进行生存分析,最终得到2个可能影响PRCC患者预后的基因,如图10所示。在Cluster1中,ZNF135低表达组患者的总生存期低于高表达组; 图10 基因ZNF135与RBPMS2的生存曲线图 本文采用rMKL-LPP方法对PRCC多组学数据进行整合分型,将PRCC患者分为三型,不同分型在通路的活性、基因表达调控方面均有差异。基于分型得到的潜在生物标记物(基因或信号通路),将为PRCC针对性的干预治疗提供重要的参考依据。 PRCC患者分为三型,Cluster1与Cluster2型PRCC发病年龄在60~65岁,且男性居多,与大多数病例的高发年龄、性别构成基本吻合[20]。预后最差的Cluster3型患者初始诊断年龄偏小,且在女性中更为常见。结合三个亚型来看,发病年龄越早的患者预后越差,而且不同性别的患者高发年龄可能不一致。对此,临床上应予以重视,多关注小于50岁的患者,同时加强对女性患者的筛查和评估,及时进行干预。本研究女性样本含量较小,有关PRCC年龄及性别的差异仍需进一步研究。 不同亚型信号通路活性的差异可帮助理解PRCC异质性的分子基础。通路TGF-β、EGFR、NF-Kβ、MAPK、Hypoxia、PI3K和TNF-α在Cluster3中活性最高。其中TGF-β可通过诱导患者上皮间质转换来促进RCC发展[21]。EGFR通路在肾脏发育过程中起关键作用,可能是PRCC一个潜在的治疗方向[22]。通路NF-Kβ和MAPK可调节RCC细胞增殖、侵袭和迁移[23-24]。Hypoxia通路作为肾癌的主要驱动因素被广泛研究,与透明细胞肾细胞癌关系密切[25-26]。PI3K在RCC中可加速细胞周期,促进肿瘤细胞迁移[27],据此,可通过抑制PI3K/AKT信号通路来阻止RCC的生长和转移[28]。TNF-α通路可为癌细胞的激活、分化、侵袭和增殖提供信号,促进癌症发展[29-30]。可见,这些通路的异常激活可能与Cluster3不良预后相关。此外,Wnt通路的异常激活可促进RCC的转移和恶化[31]。而VEGF和Estrogen通路可影响肾癌的发生发展[32-33],其对于PRCC靶向治疗的意义有待进一步挖掘。 本研究基于PRCC分子分型得到三个可能受DNA甲基化或miRNA调控且影响患者生存的基因ABL2、ZNF135与RBPMS2,这三个差异分子靶标与PRCC的关系尚不明确,但有研究发现它们与其他癌症有关。ABL2是一种原癌基因,可参与调控细胞生长、侵袭和迁移等过程[34],与肾细胞癌的发生发展相关[35]。ZNF135编码一种转录抑制蛋白,在透明细胞肾细胞癌、宫颈癌与乳腺癌等多种癌症中高度甲基化[36],本研究发现其在RPCC中也高度甲基化,具体作用机制有待进一步阐明。RBPMS2的高表达与胃肠道间质瘤有密切联系[37],还可促进乳腺癌的发生发展[38]。这些基因对PRCC的预后有一定的预测价值,有望作为PRCC未来药物治疗的潜在靶点,而且基因间的调控作用也值得进一步研究。 综上所述,本文基于rMKL-LPP方法对PRCC多组学数据的整合分析,能够有效地识别亚型,为PRCC的分型研究提供了新的思路。识别出的PRCC亚型在信号通路活性、基因表达及调控方面均存在差异,这有助于进一步理解不同分型发生发展的潜在分子机制。此外,筛选出的潜在生物标志物将为PRCC治疗和预后评估提供一定的理论依据和临床指导。



病理分期差异有统计学意义,Ⅳ期患者死亡风险是Ⅰ期患者的20.351倍。
通路Wnt和VEGF在Cluster2中活性最高,Estrogen通路在Cluster1中活性最高。不同分型通路活性的差异也在一定程度上反映了PRCC不同亚型的异质性。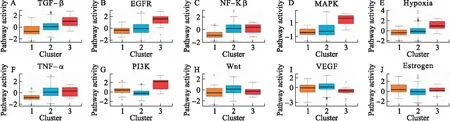
459个差异甲基化位点映射到416个DMGs,包括111个高甲基化基因和305个低甲基化基因;
筛选出13个DEmiRNAs,其中2个上调,11个下调。图5依次为DEmRNAs,DMGs及DEmiRNAs表达热图,从图中可以明显看出这些特征在不同亚型中的表达差异。



而RBPMS2仅在Cluster3中存在相关关系。对DEmRNAs与DEmiRNAs靶基因的重合基因进行相关性分析,发现ABL2仅在Cluster2中与hsa-miR-107存在负相关关系(图9)。基因之间的相关关系表明基因间可能存在生理学调控作用。
在Cluster3中,RBPMS2高表达的患者较低表达的患者预后更差。